|
|
|
|
|
|
|
Jean-Christophe Nebel
|
|
 |
Structure-based description of binding sites
Duration: 2003-2004
|
| Project description |
The simultaneous alignment of several protein sequences is now an essential step in protein analysis. It allows the detection of distant homologues and the identification of patterns from which protein activities can be inferred.
With the increasing availability of protein 3D structures, the generation of biologically meaningful 3D patterns or 3D motifs from the simultaneous alignment of several protein structures becomes an exciting prospect.
Applications:
|
|
|
|
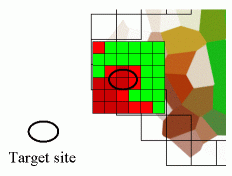
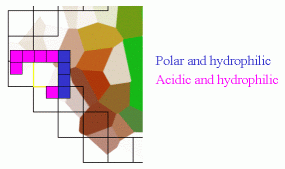
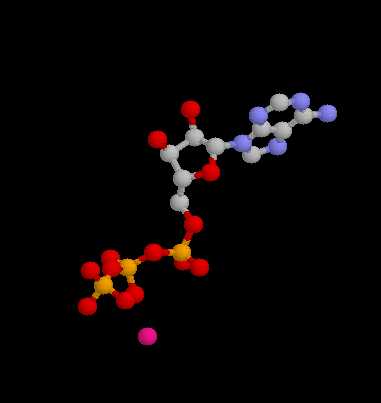
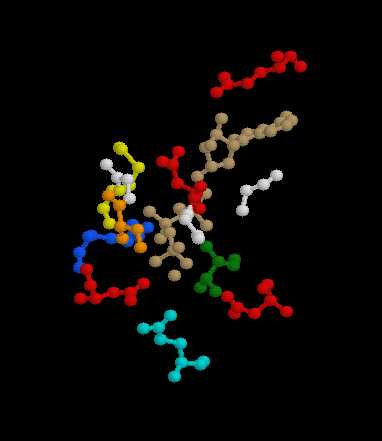
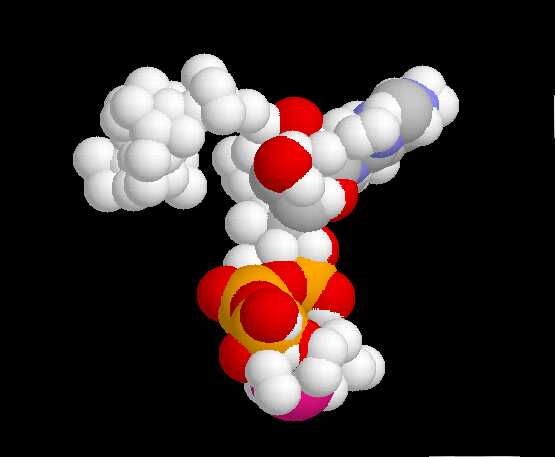
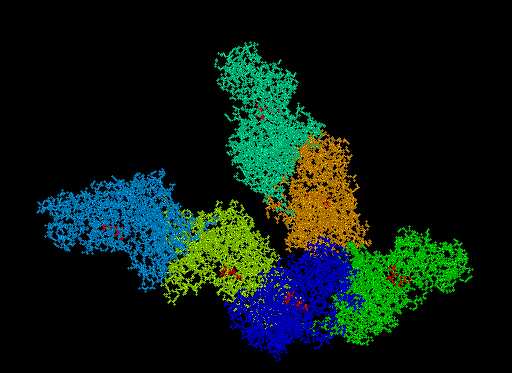
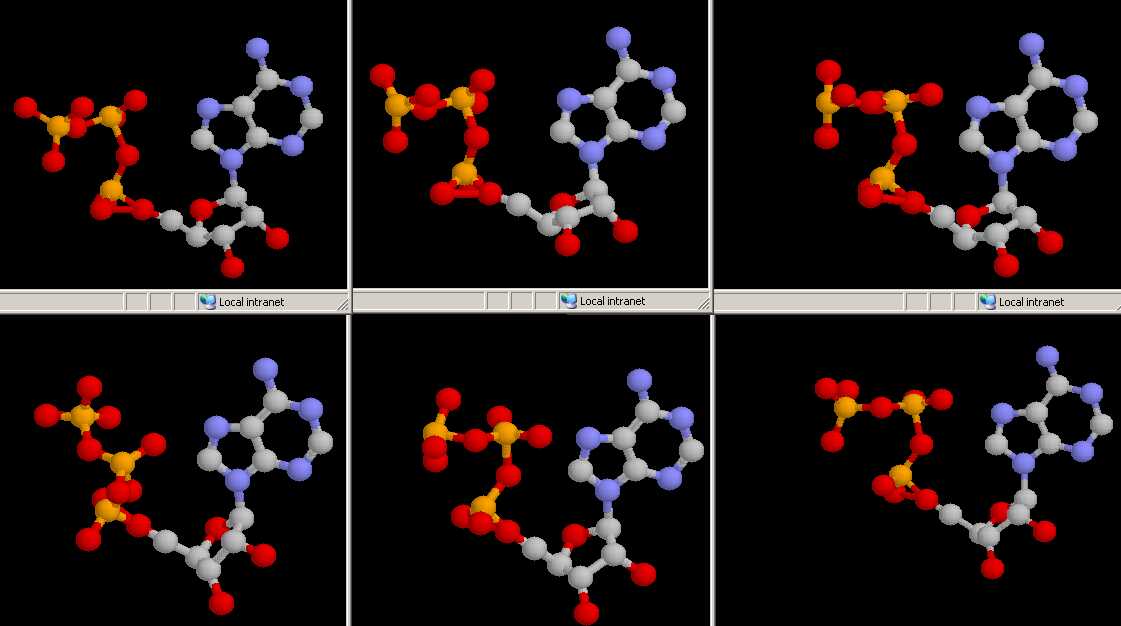
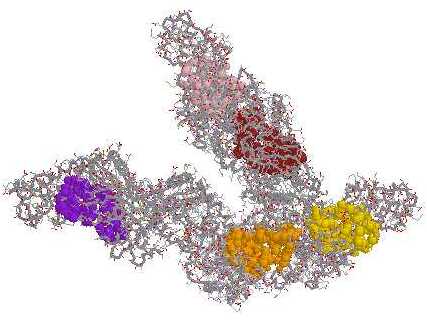
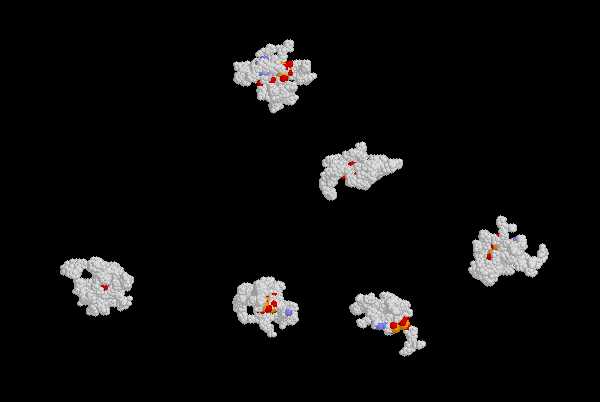
| Pilot Study: Description of ATP binding sites |
In collaboration with: David Gilbert (Professor of Bioinformatics) and Pawel Herzyk (Biochemistry)
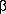 -actin, 2BTF.pdb)
-actin, 2BTF.pdb)
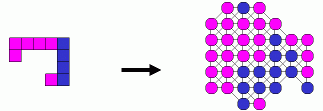
Our algorithm is based on:
|
|
||||
|
|
||||
|
|
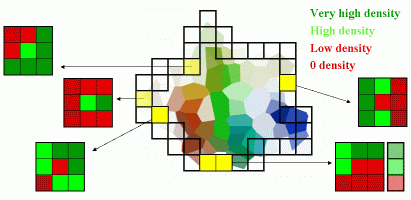
Geometry is expressed by the volume of the cavity and the values of its principal axes.
|
|
||||
|
|
| Research interest keywords |
Structural Bioinformatics, 3D Graphics, 3D Structure, Binding Site, Protein Docking